
Well-Tempered Metadynamics Simulation
The Well-Tempered Metadynamics method is an enhanced sampling method described in the Theory section. It adds external energy for pushing the system to explore different
conformations. to avoid atom dissociation, we add a restraint to the system. This restriction consists of a
semi-harmonic potential with the form
where $k$ is a constraint, in this case being 1000; $r_w$ is the location of the constraint, that we choose to be 2 (in LJ dimensionless reduced units); $d_i$ is the distance of each atom to the center of mass. Note that this potential does not do anything if the distance between the atom and the center of mass is lower than $r_w$, but if it is greater this potential begins to exert a force over the atoms and to avoid dissociation. This is defined with the keyword UPPER_WALLS in the plumed setup.
An example PLUMED file with the information of the constraints (also called walls) and the coordinates you want to bias can be found here (plumedMTD-LJ.dat):
UNITSThis command sets the internal units for the code. More details LENGTHthe units of lengths=A TIMEthe units of time=0.0101805 ENERGYthe units of energy=96.4853329 COMCalculate the center of mass for a group of atoms. More details ATOMSthe list of atoms which are involved the virtual atom's definition=1-7 LABELa label for the action so that its output can be referenced in the input to other actions=com DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=1,com LABELa label for the action so that its output can be referenced in the input to other actions=d1 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d1 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=2,com LABELa label for the action so that its output can be referenced in the input to other actions=d2 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d2 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=3,com LABELa label for the action so that its output can be referenced in the input to other actions=d3 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d3 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=4,com LABELa label for the action so that its output can be referenced in the input to other actions=d4 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d4 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=5,com LABELa label for the action so that its output can be referenced in the input to other actions=d5 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d5 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=6,com LABELa label for the action so that its output can be referenced in the input to other actions=d6 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d6 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. DISTANCECalculate the distance/s between pairs of atoms. More details ATOMSthe pair of atom that we are calculating the distance between=7,com LABELa label for the action so that its output can be referenced in the input to other actions=d7 UPPER_WALLSDefines a wall for the value of one or more collective variables, More details ARGthe arguments on which the bias is acting=d7 ATthe positions of the wall=2.0 KAPPAthe force constant for the wall=100. c1: COORDINATIONNUMBERCalculate the coordination numbers of atoms so that you can then calculate functions of the distribution of This action is a shortcut. More details SPECIESthe list of atoms for which the symmetry function is being calculated and the atoms that can be in the environments=1-7 MOMENTSthe list of moments that you would like to calculate=2-3 SWITCHthe switching function that it used in the construction of the contact matrix. Options for this keyword are explained in the documentation for LESS_THAN.={RATIONAL R_0=1.5 NN=8 MM=16} METADUsed to performed metadynamics on one or more collective variables. More details ARGthe labels of the scalars on which the bias will act=c1.* HEIGHTthe heights of the Gaussian hills=0.05 PACEthe frequency for hill addition=500 SIGMAthe widths of the Gaussian hills=0.1,0.1 GRID_MINthe lower bounds for the grid=-1.5,-1.5 GRID_MAXthe upper bounds for the grid=2.5,2.5 GRID_BINthe number of bins for the grid=500,500 BIASFACTORuse well tempered metadynamics and use this bias factor=5 FILE a file in which the list of added hills is stored=HILLS
The Well-Tempered Metadynamics simulation can be run using the code
MTD.py:
from ase.calculators.lj import LennardJones
from ase.calculators.plumed import Plumed
from ase.constraints import FixedPlane
from ase.md.langevin import Langevin
from ase.io import read
from ase import units
timestep = 0.005
ps = 1000 * units.fs
setup = open("plumedMTD-LJ.dat", "r").read().splitlines()
atoms = read('isomerLJ.xyz')
cons = [FixedPlane(i, [0, 0, 1]) for i in range(7)]
atoms.set_constraint(cons)
atoms.set_masses([1, 1, 1, 1, 1, 1, 1])
atoms.calc = Plumed(calc=LennardJones(rc=3., r0=2.5, smooth=True),
input=setup,
timestep=timestep,
atoms=atoms,
kT=0.1)
dyn = Langevin(atoms, timestep, temperature_K=0.1/units.kB, friction=1,
fixcm=False, trajectory='MTD.traj')
dyn.run(500000)
Note that Well-Tempered Metadynamics requires the value of the temperature according to equation (2) . Then, it is necessary to define the kT argument of the calculator. SIGMA and PACE are the standard deviation of the Gaussians and the deposition interval in terms of the number of steps ($\tau$ in equation (1)), respectively. HEIGHT and BIASFACTOR are the maximum height of the Gaussians (W) and the $\gamma$ factor of equation (2), respectively.
In this case, the Lennard-Jones calculator computes the forces between atoms, namely, ${\bf F}_i$ forces in equation (3). Likewise, you can use any preferred ASE calculator in place of the LJ calculator used here.
When one runs a metadynamics simulation, Plumed generates an output file called HILLS that contains the information of the deposited Gaussians. You can reconstruct the free energy by yourself or can use the plumed tool sum_hills. The simplest way of using it is:
$ plumed sum_hills --hills HILLS
After this, Plumed creates a fes.dat file with the free energy surface (FES) reconstructed. The FES of this example is plotted using plotterFES.py:
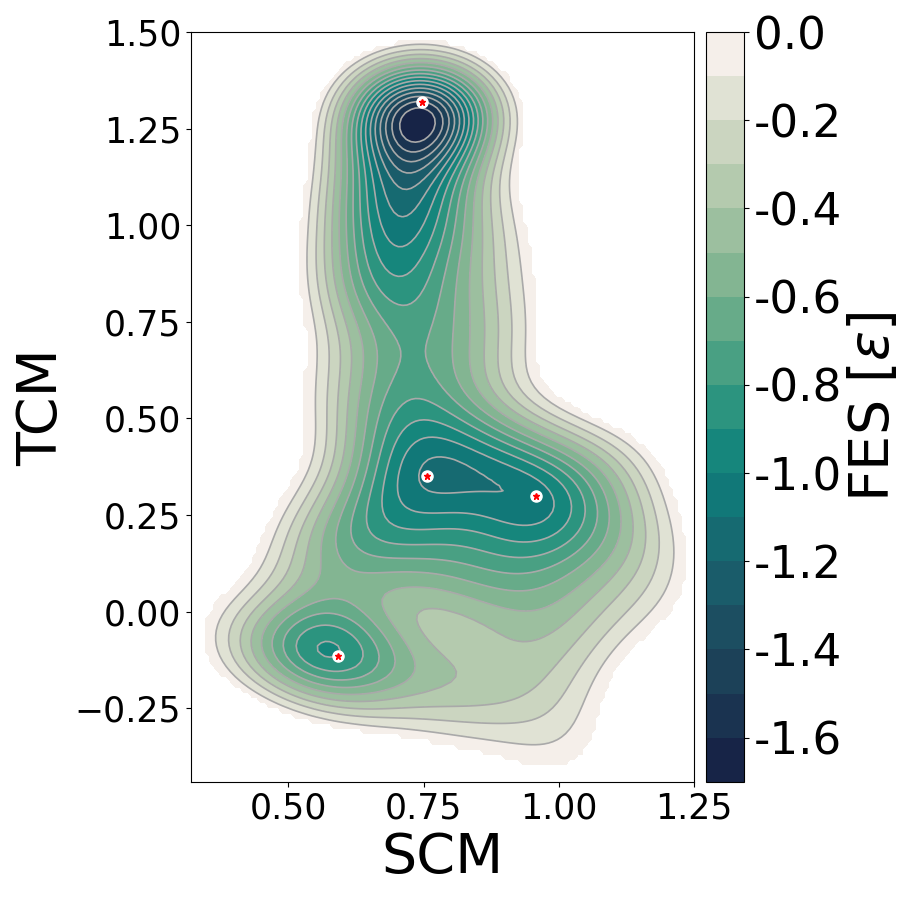
Figure 3. Free energy surface (LJ units) in the space of the collective variables second and third central moment. Orange stars represent the location of the local minima isomers of the LJ cluster in this space.
Note that bias is added over all states, meaning that the system jumped from one state to another and gave a complete reconstruction of the free energy surface.